
Postmarketing Adverse Drug Experience (PADE) Inspections – Part II
Legal Framework of PADE Inspections
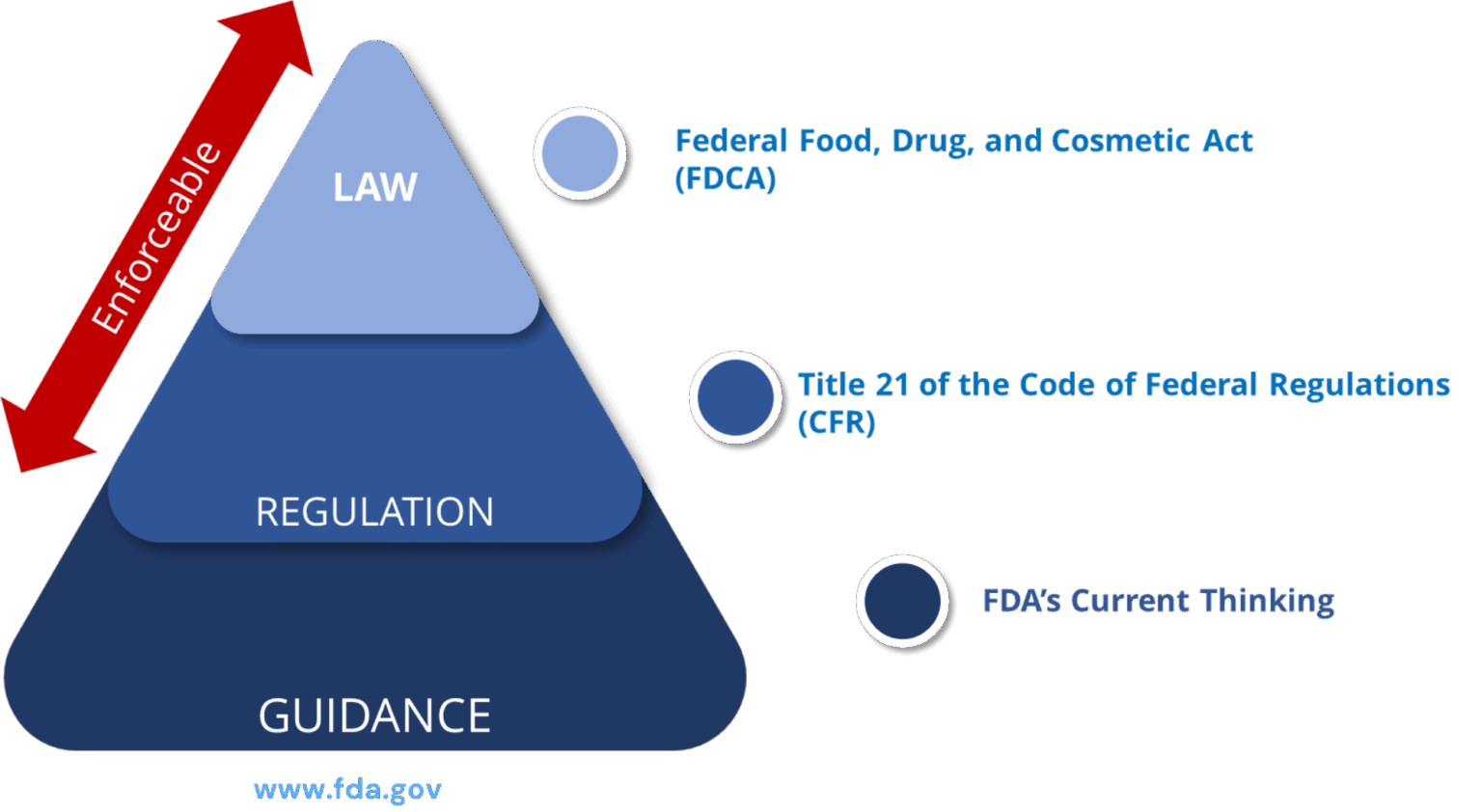
- LAW: Federal Food, Drug and Cosmetic Act (FDCA)
- Title 21 of the Code of Federal Regulations (CFR)
- FDA’s Current Thinking
Inspectional Observations: USFDA 2023
| 1 | 21 CFR 314.80(b) | Failure to develop written procedures | Written procedures have not been developed for the [surveillance] [receipt] [evaluation] [reporting to FDA] of post marketing adverse drug experiences. |
| 2 | 21 CFR 314.81(b)(1)(ii) | Failure to meet specifications | An NDA-Field Alert Report was not submitted within three working days of receipt of information concerning a failure of one or more distributed batches of a drug to meet the specifications established for it in the application. |
| 3 | 21 CFR 314.80(c)(1)(i) | Late submission of 15-day report. | Not all adverse drug experiences that are both serious and unexpected have been reported to FDA within 15 calendar days of initial receipt of the information. |
| 4 | 21 CFR 314.80(c)(1)(ii) | Failure to investigate serious, unexpected events | Adverse drug experiences that were the subject of post marketing 15-day reports were not [promptly] investigated. |
| 5 | 21 CFR 314.80(c)(2) | Late submission of annual safety reports | Not all annual periodic adverse drug experience reports have been submitted within 60 days of the anniversary date of the approval of the application. |
| 6 | 21 CFR 314.80(c)(2)(ii)(A) | Incomplete periodic safety report | Failed to submit a periodic report containing
|
| 7 | 21 CFR 314.80(d) | Failure to submit scientific article | A postmarketing 15-day Alert report based upon scientific literature was not accompanied by a copy of the published article. |
| 8 | 21 CFR 314.80(j) | Failure to maintain records | Failed to maintain for a period of 10 years records of all adverse drug experiences known to you, including raw data and any correspondence. |
| 9 | 21 CFR 314.81(b)(2) | Timely submission | An annual report was not submitted [each year] [within 60 days of the anniversary date of U.S. approval of the application] to the FDA division responsible for reviewing the application. |
Ref: Number of 483 issued from the System*
Inspections ending between 10/1/2022 and 9/30/2023
https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/inspection-observations
Written Procedures Must Address
- Surveillance
- Account for all sources
- Spontaneous
- Solicited
- Internet sources (firm sponsored)
- Literature …and more!
- Receipt
- ADE info
- Initial
- Follow-up
- Receipt from any source
- Evaluation
- Seriousness
- Expectedness
- Relatedness
- ADEs from any source
- Follow-up procedures
- Reporting
- 15-day Alert Reports
- Non-expedited individual case safety reports (ICSRs)
- Aggregate Reports
- All info must be submitted electronically
Ref: Postmarketing Drug Safety and Inspection Readiness June 19, 2018. Center for Drug Evaluation and Research (CDER) Small Business and Industry Assistance (SBIA) Webinar. USFDA
Written Procedures
- Develop written procedures for the surveillance, receipt, evaluation, and
reporting of postmarketing safety information, including procedures for
managing safety information with contractors and business partners, as
applicable. - Written procedures should be maintained and followed.
- Determine if written procedures provide for complete, accurate, and timely reporting of safety data to FDA.
- Regulations pertaining to the requirements for written procedures: 21 CFR 4 310.305(a), 21 CFR 314.80(b), and 21 CFR 600.80(b).
Surveillance
- Determine if the firm is monitoring potential sources of adverse event information
- Determine if the firm is surveilling both foreign and domestic sources.
- Determine if the firm is promptly reviewing all postmarketing safety information received from any source
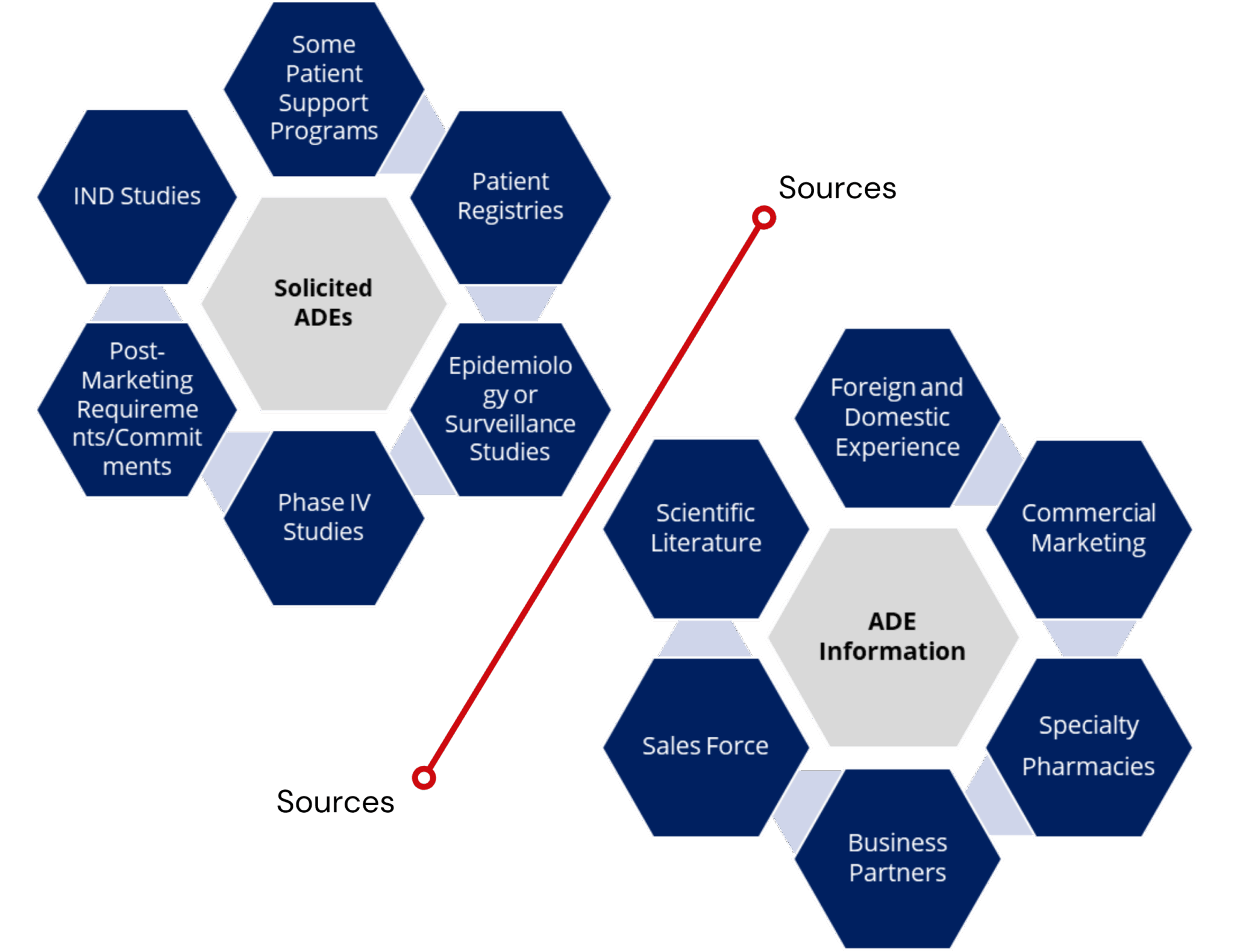
Many other sources such as firm-sponsored websites, firmsponsored social media, legal cases, product complaint files etc.
- The timeline for submission of adverse experiences to FDA begins the day that the applicant, nonapplicant, or its contractors or business partners, obtain the minimum data set for a valid adverse event report.
- The minimum dataset required to consider information reportable is
- an identifiable patient
- an identifiable reporter
- a suspect product and
- an event.
- The date of receipt must be accurately determined and documented for the receipt of initial and follow-up information received by any method (for example, by phone, electronic mail, postal mail, fax, literature, websites, or employees).
Evaluation
Determine how:
- Safety information from any source is evaluated to determine if an adverse experience is present.
- Adverse experience reports are evaluated to establish if each report is spontaneous or solicited.
- All adverse experiences, both spontaneous and solicited, are evaluated for seriousness and expectedness.
- For adverse experiences originating from solicited sources, determine how the causal relationship between the product and the adverse experience is assessed.
- If adverse experiences that are both serious and unexpected are promptly investigated and if all attempts to obtain additional information are documented.
Reporting
- Spontaneous adverse experiences, foreign or domestic, that have been evaluated as both serious and unexpected are submitted to FDA no later than 15 calendar days from the initial receipt of the information.
- For solicited adverse experiences, foreign or domestic, determine if all adverse experiences that have been evaluated as serious, unexpected, and possibly related to the suspect product are submitted to FDA no later than 15 calendar days from initial receipt of the information.
- Review 15-day Alert reports submitted late to the Agency.
- For each late report, the firm should provide justification for why the reports were late and appropriate corrective actions, if applicable.
- Domestic spontaneously reported non-expedited ICSRs are being submitted to FDA with or before the Periodic Report.
- The firm is in possession of any adverse event data that were not reported to the Agency as required.
Stay tuned for Part III, where we will explore the safety reports.
References
- https://www.fda.gov/
- Number of 483 issued from the System* Inspections ending between 10/1/2022 and 9/30/2023 https://www.fda.gov/inspections-compliance-enforcement-and- criminal-investigations/inspection-references/inspection-observations
- Postmarketing Drug Safety Compliance: 2019 Inspection Findings April 29, 2020 (Live Webinar) Center for Drug Evaluation and Research – Small Business and Industry Assistance, Center for Drug Evaluation and Research, US Food and Drug Administration
- Postmarketing Drug Safety and Inspection Readiness June 19, 2018 Center for Drug Evaluation and Research (CDER) Small Business and Industry Assistance (SBIA) Webinar
- Postmarketing Drug Safety and Inspection Readiness June 19, 2018 Center for Drug Evaluation and Research (CDER) Small Business and Industry Assistance (SBIA) Webinar
- CHAPTER 53 – Postmarketing Surveillance and Epidemiology: Human Drug and Therapeutic Biological Products, fda.gov
About Soterius
Soterius is a strong team of pharma professionals who design customized, innovative, and costefficient processes for clinical safety, pharmacovigilance, and medical affairs. Our deep industry knowledge and up to date insights let us combine agile, people powered intelligence in pioneering customer centric solutions. Our innovative technology solutions include engagement tools and communications platforms to create a unified and compliant medical access facility. With a strong global presence, we provide comprehensive clinical and post
marketed safety services, that include aggregate report writing, signal detection and management, global literature surveillance, risk management, case processing and regulatory reporting.
We use state-of-the-art technologies to solve complex safety operations problems, be it case processing, intake, site reporting for clinical trials, or literature search and management. We have one of the most accurate solutions for case intake and case processing using AI. Like This Blog?
We support companies from the initial development stage of a drug/vaccine to the approval and ultimate marketing of the therapy, supporting ongoing operations and regulatory commitments globally.
Authors

Dr. Sumit Verma MD, DNB
President, Clinical Safety and PV
Dr. Sumit Verma is a medical graduate with specialization in anesthesiology and has more than 15 years of experience in the pharmaceutical industry, clinical medicine, clinical research, and pharmacovigilance. He has built teams that have consistently delivered and exceeded customer expectations across pharmacovigilance domains such as case processing, signal management, risk management, aggregate reports, and clinical safety. He has co-authored two books – one on pharmacovigilance and another on pharmacology.