
Signal Detection in Early Phase Clinical Drug Trials
Why Should You Perform Signal Detection in Early Phase Clinical Drug Trials?
The term ‘pharmacovigilance’ has conventionally been related with post-marketing activities; however, it is also equally applicable to the pre-marketing process for collecting, managing & assessing safety information during clinical development of the molecule. Similarly, the concepts of signal detection and assessment, risk assessment and risk minimization are as applicable to the pre-marketing scenario as they are to the post-marketing scenario.
The importance of this is evident from the following examples:
-
Drugs not approved in USA
– Examples of drugs not approved in USA as premarketing experience provided evidence of the potential for severe DILI e.g., Dilevalol, Tasosartan, Ximelagatran.
-
Drugs withdrawn from market worldwide after initial regulatory approval
– E.g., Rimonabant, Rofecoxib, celecoxib etc.
The safety information generated at the end of clinical development program should be extensive enough to permit comprehensive regulatory review and determination of benefit-risk profile for supporting marketing approval. The information should be comprehensive so that product label can be adequate to provide prescribers & patients adequate information for safe use of the drug.
What Do the Regulators Say?
USFDA 21CFR312.32 USFDA Draft Guidance on Sponsor Responsibilities
June 2021
Sponsors must adopt a systematic approach to safety surveillance to meet IND safety reporting requirements and enhance quality of safety reporting.
Such an approach involves promptly reviewing, evaluating, and managing safety information from all sources, including animal studies, clinical investigations, scientific literature, professional meetings, foreign regulatory authorities, and commercial marketing experience.
USFDA 21CFR312.56 USFDA Draft Guidance on Sponsor Responsibilities June 2021
– The sponsor’s review should involve assessing data from all sources and monitoring the progress of investigations:
- To identify previously undetected potential serious risks (§312.56(a)).
- To update investigator’s brochure, protocol, and consent forms with new information.
- As necessary, to take measures for protecting subjects (e.g. monitoring, modifying dosing, or participant selection) (§312.56(d)).
CIOMS Working Group VI
– The purpose of ongoing safety evaluation during drug development is to ensure that important safety signals are detected early and to have a better understanding of benefit-risk profile of study drug.
Sponsors should develop a system to analyse, evaluate and take actions on the safety information on a continuous basis. This is to ensure the earliest possible detection of safety concerns and allow suitable risk minimization.
Systemic Approach for Signal Detection

Essential Elements for Effective Signal Detection in a Clinical Development Program
- Early Initiation is Crucial
- Written Process Required for Review
- Multidisciplinary Safety Management Team (SMT)
- Data from Licensing Partners to be Considered
- Project Management Function
- Background Incidence
- Ready Data Accessibility
- Initiative Proactive Strategy
- Establish Timeframes and Milestones
- Decision Making
- Advisory Bodies
Signal Detection: Effective Data Review
Below are some of the key points that support effective data review during signal detection process in a clinical development program:
| Parameters | Overview |
| Analyse all AEs – Serious and Non-serious | The safety analysis is complete when all adverse events (serious and non-serious) are reviewed, with a greater emphasis on those leading to treatment discontinuation |
| Review of Individual Cases | Individual case analysis is essential for safety analysis. SARs and AESIs are important for detecting safety signals. Consider patient population, drug indication, and disease history during analysis. |
| Aggregate Review of Safety Data | Aggregate review helps analyse evolving safety information, comparing interval and cumulative data. Data should be analysed per dose, cohort, gender, age, etc. |
| Review of Clinical Lab Data | Laboratory tests are useful for screening subjects, early detection of organ toxicity & detection of potential toxic effects. Special focus should be on lab values correlating with organ toxicity e.g., endocrine abnormalities, hepatotoxicity etc. |
| Adverse Events of Special Interest (AESI) | The protocol should define AESIs, emphasizing identification, monitoring, and reporting. Includes events like rhabdomyolysis or hair loss. |
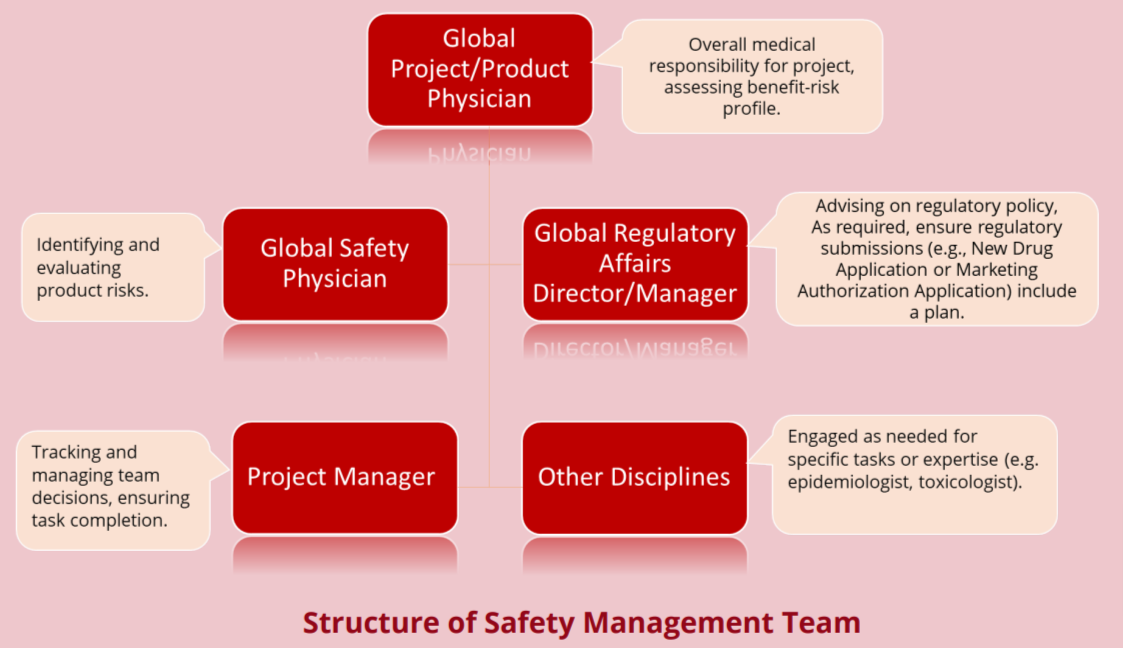
Structure of Safety Management Team
Development Risk Management Plan (DRMP)
A Development Risk Management Plan (DRMP) is a natural extension of high-quality pharmacovigilance. In DRMP, a compound-specific approach should be adopted, possibly as part of the broader Clinical Development Plan. It should contain early documentation of identified, expected, or potential risks, along with strategies for addressing them throughout development. As appropriate, the DRMP may develop into a post-marketing risk management plan.
The DRMP is a guide for safety surveillance during development and is not a legal or regulatory document; however, the following two actions must be considered during development of the process:
Recognize the potential for legal discovery of DRMP and ensure appropriate
language clarifying its status as a working document.
- Recognize the potential for legal discovery of DRMP and ensure appropriate language clarifying its status as a working document.
- Establish robust processes, including project management to ensure the diligent execution of the action plans.
The DRMP should include the following sections: anticipated product profile, epidemiology, non-clinical safety experience, clinical safety experience, identification and assessment of known or anticipated risks, identification and assessment of potential new risk, and actions and/or plans for evaluating and mitigating risk.
Conclusion
The concepts of signal assessment, risk assessment and risk minimization are as
applicable to pre-marketing scenario as they are to the post-marketing scenario. The purpose of ongoing safety evaluation is to ensure that safety signals are detected early and to obtain an understanding of benefit-risk profile of the drug. Signal detection during clinical trials is usually performed based on clinical judgement, since there is limited data available during premarketing clinical trials. The three basic attributes for signal detection includes quick medical assessment, periodic aggregate assessment, and safety evaluation of completed unblinded trials. To ensure effective signal management, it is important to establish an effective system, beginning early, having proactive approach, analysing all serious and non-serious events, periodic reviews by scientific committees, and prompt decision making.
References
- Report of CIOMS Working Group VI: Management of Safety Information from Clinical Trials: Geneva 2005.
- Report of CIOMS Working Group VIII: Practical Aspects of Signal Detection in Pharmacovigilance: Geneva 2010.
- USFDA Guidance for Industry: Sponsor Responsibilities – Safety Reporting Requirements and Safety Assessment for IND and Bioavailability/Bioequivalence Studies: June 2021.
- 21CFR312.32: Subpart B – Investigational New Drug Application (IND): IND safety reporting.
- 21CFR312.56: Subpart D – Responsibilities of Sponsors and Investigators: Review of ongoing investigations.
- European Medicines Agency: Guideline on good pharmacovigilance practices (GVP) Module IX – Signal management (Rev 1); Dated 22 November 2017.
About Soterius
Soterius is a strong team of pharma professionals who design customized, innovative, and cost-efficient processes for clinical safety, pharmacovigilance, and medical affairs. Our deep industry knowledge and up to date insights let us combine agile, people powered intelligence in pioneering customer centric solutions. Our innovative technology solutions include engagement tools and communications platforms to create a unified and compliant medical access facility. With a strong global presence, we provide comprehensive clinical and post marketed safety services, that include aggregate report writing, signal detection and management, global literature surveillance, risk management, case processing and regulatory reporting. We use state-of-the-art technologies to solve complex safety operations problems, be it case processing, intake, site reporting for clinical trials, or literature search and management. We have one of the most accurate solutions for case intake and case processing using AI.
We support companies from the initial development stage of a drug/vaccine to the approval and ultimate marketing of the therapy, supporting ongoing operations and regulatory commitments globally.
Authors

Dr. Sumit Verma MD, DNB
President, Clinical Safety and PV
Dr. Sumit Verma is a medical graduate with specialization in anesthesiology and has more than 15 years of experience in the pharmaceutical industry, clinical medicine, clinical research, and pharmacovigilance. He has built teams that have consistently delivered and exceeded customer expectations across pharmacovigilance domains such as case processing, signal management, risk management, aggregate reports, and clinical safety. He has co-authored two books – one on pharmacovigilance and another on pharmacology.

Dr. Yogesh Gulati MD
Sr. Safety Physician, Clinical Safety and PV
Dr. Yogesh Gulati is a medical graduate with specialization in pharmacology and has more than 13 years of experience in the pharmaceutical industry, clinical research, and various phases of clinical trials. He has led various pharmacovigilance teams comprising of physicians and clinical research coordinators in conducting pharmacovigilance activities for various global clients. He has been involved in setup of a standalone pharmacovigilance unit and gradual scale up of operations while ensuring system and regulatory compliance. He has led teams that have delivered quality documents across various pharmacovigilance domains including case processing, signal management, risk management, and aggregate reports. He has co-authored books on pharmacovigilance, pharmacology and nursing drug guide.
Disclaimer
Copyright 2025 by Soterius, Inc. All rights reserved. Soterius logo are trademarks or registered trademarks of Soterius in all jurisdictions. Other marks may be trademarks or registered trademarks of their respective owners. The information you see, hear or read on the pages within this presentation, as well as the presentation’s form and substance, are subject to copyright protection. In no event, may you use, distribute, copy, reproduce, modify, distort, or transmit the information or any of its elements, such as text, images or concepts, without the prior written permission of Soterius. No license or right pertaining to any of these trademarks shall be granted without the written permission of Soterius (and any of its global offices and/or affiliates). Soterius reserves the right to legally enforce any infringement of its intellectual property, copyright and trademark rights. Any content presented herewith should only be considered for general informational purposes and should not be considered as specific to the requirements of any particular organisation or for any specific purpose. Soterius does not make any representations or warranties about the completeness, reliability, appropriateness, relevance, or accuracy of the content presented here.
Confidential Information
Copyright@2025
Soterius, Inc
Discover more
 abinash kumar •
10 Min Read
abinash kumar •
10 Min Read
Governance and Accountability in AI-Enabled Pharmacovigilance
Governance and accountability are foundational to the use of artificial intelligence in pharmacovigilance. AI systems do not exist outside the pharmacovigilance system in which they are deployed. Decisions about their…
 abinash kumar •
10 Min Read
abinash kumar •
10 Min Read
Human Oversight in AI-Enabled Pharmacovigilance: What ‘HITL’ Actually Has to Mean
1. Why Human Oversight Is a Control, Not a Reassurance As artificial intelligence becomes embedded in pharmacovigilance workflows, the presence of a human reviewer is often assumed to be a…
 admin •
10 Min Read
admin •
10 Min Read
Deploying an Inspection Ready AI System in PV
Artificial intelligence in pharmacovigilance should not be assessed by how advanced the technology is. It should be assessed by what happens when the technology is wrong. Executive Summary Artificial intelligence…