
Postmarketing Adverse Drug Experience (PADE) Inspections – Part IV
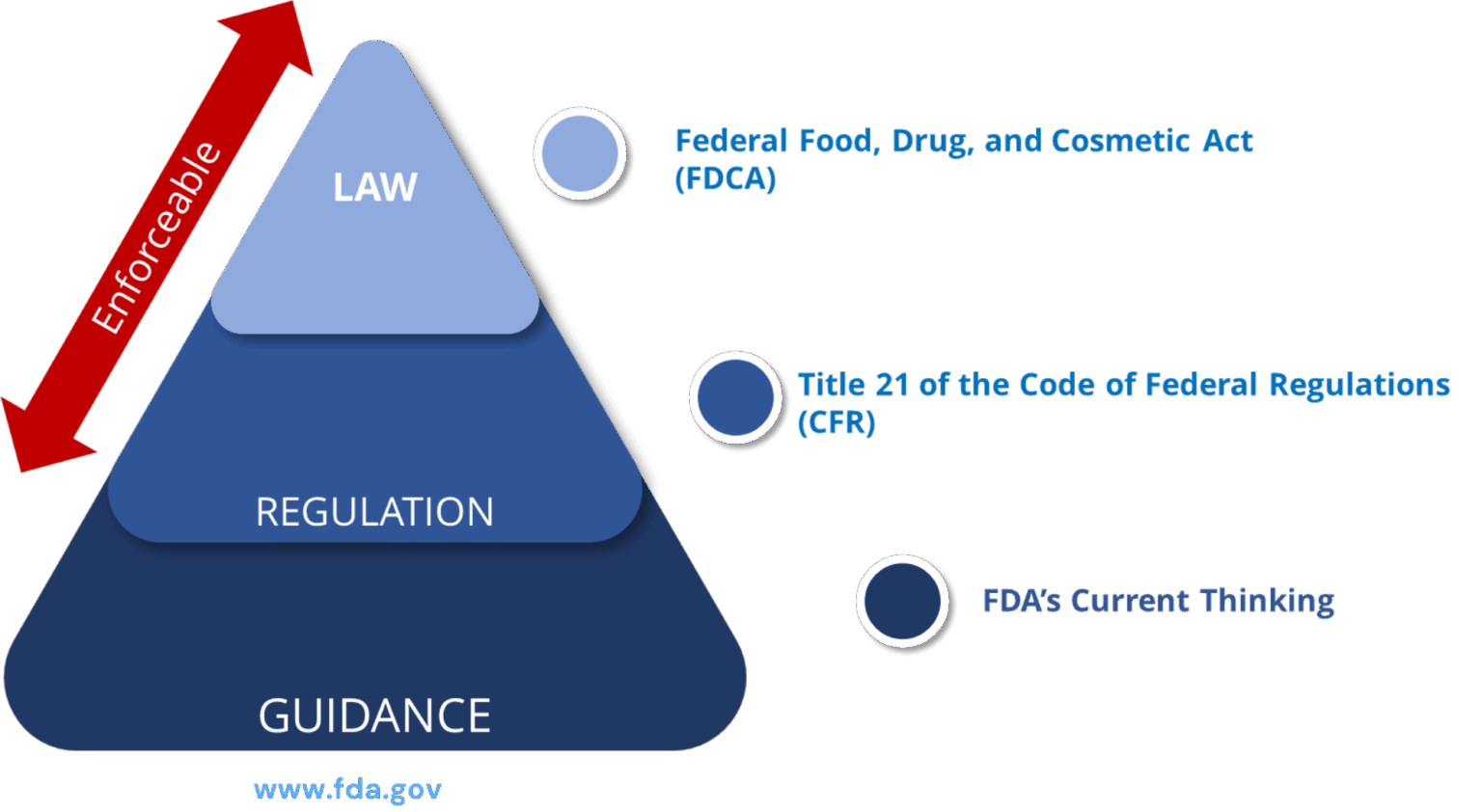
Legal Framework of PADE Inspections
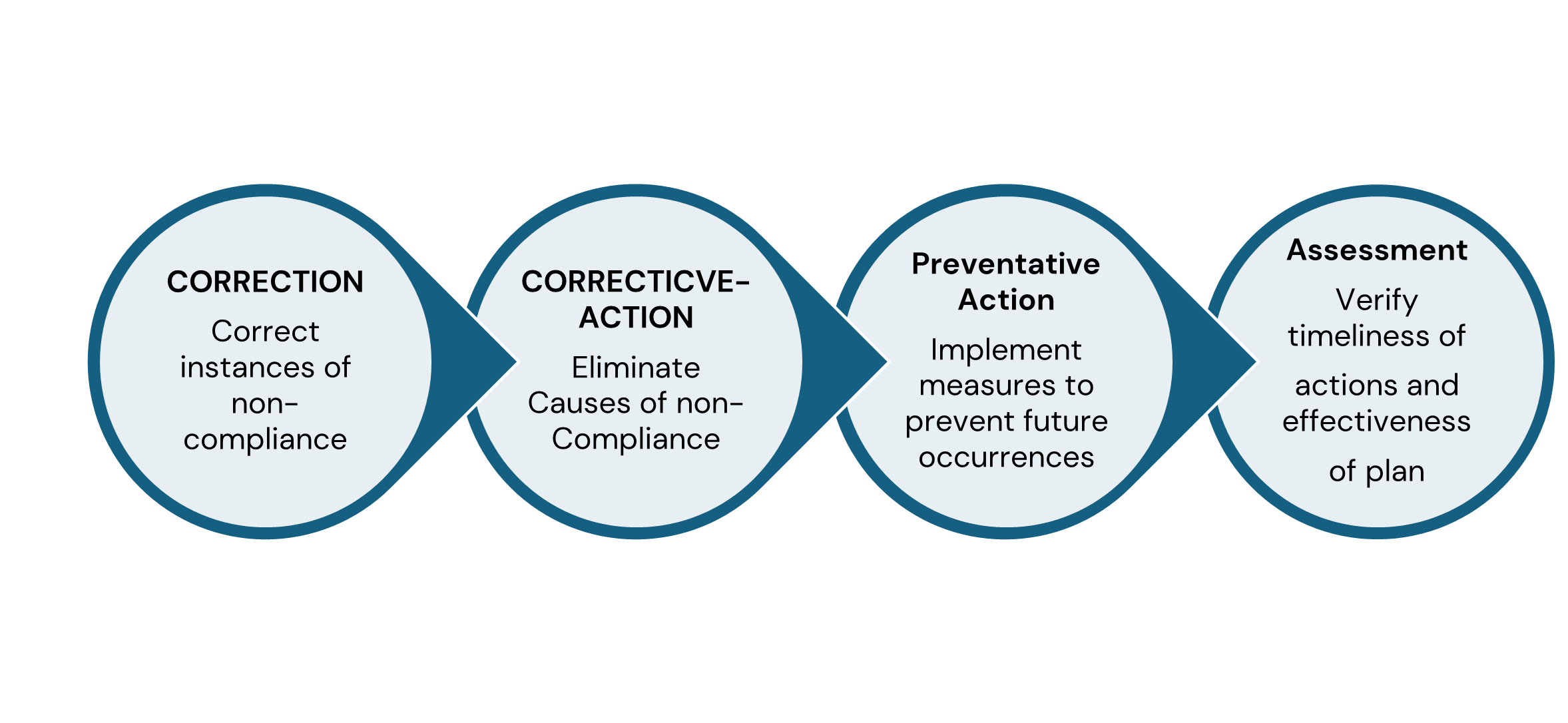
Good Corrective Action Plan – Four Reasons to Submit a Complete and Timely
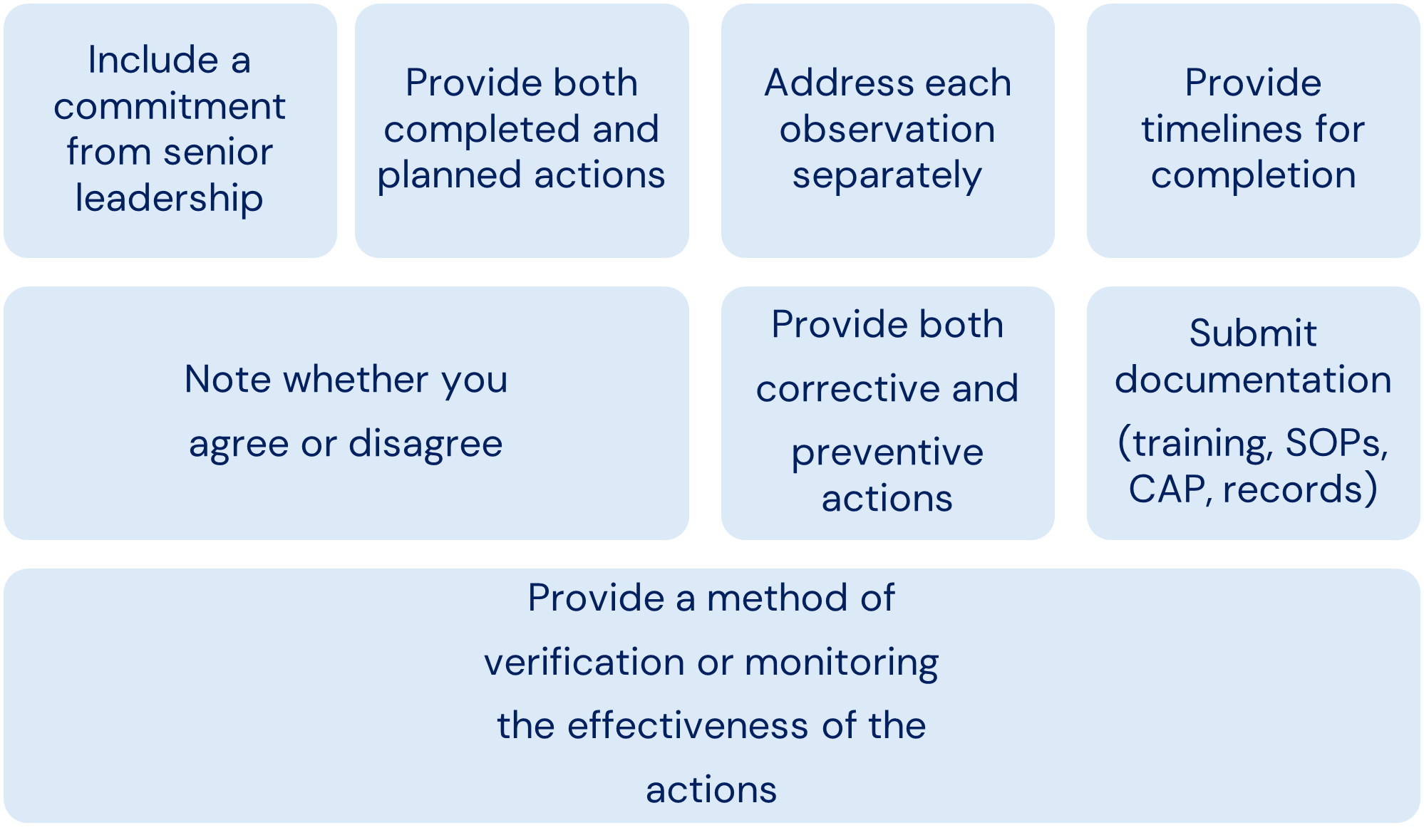
Written Response
- May be considered in an FDA compliance decision.
- Demonstrates your acknowledgment and understanding of the observations to the FDA
- Demonstrates your commitment to correct the observations to the FDA
- Establishes credibility with the FDA
Points to Consider for Written Responses to the FDA
(SUBMIT THE REPORT WITHIN 15 WORKING DAYS)
Inspection Reporting: FORM FDA 483, Inspectional Observations

Inspection Classifications
| No Actions Indicated
(NAI) |
Voluntary Action Indicated (VAI) | Official Action Indicated (OAI) |
| No Objectionable conditions
or practices were found during an inspection (or the objectionable conditions found do not justify further regulatory action).
|
Objectionable conditions or practices were found, but do not rise to the level warranting OAI classification. | Objectionable conditions or practices were found, whose scope, severity, or pattern warrants the recommendation for a regulatory action. |
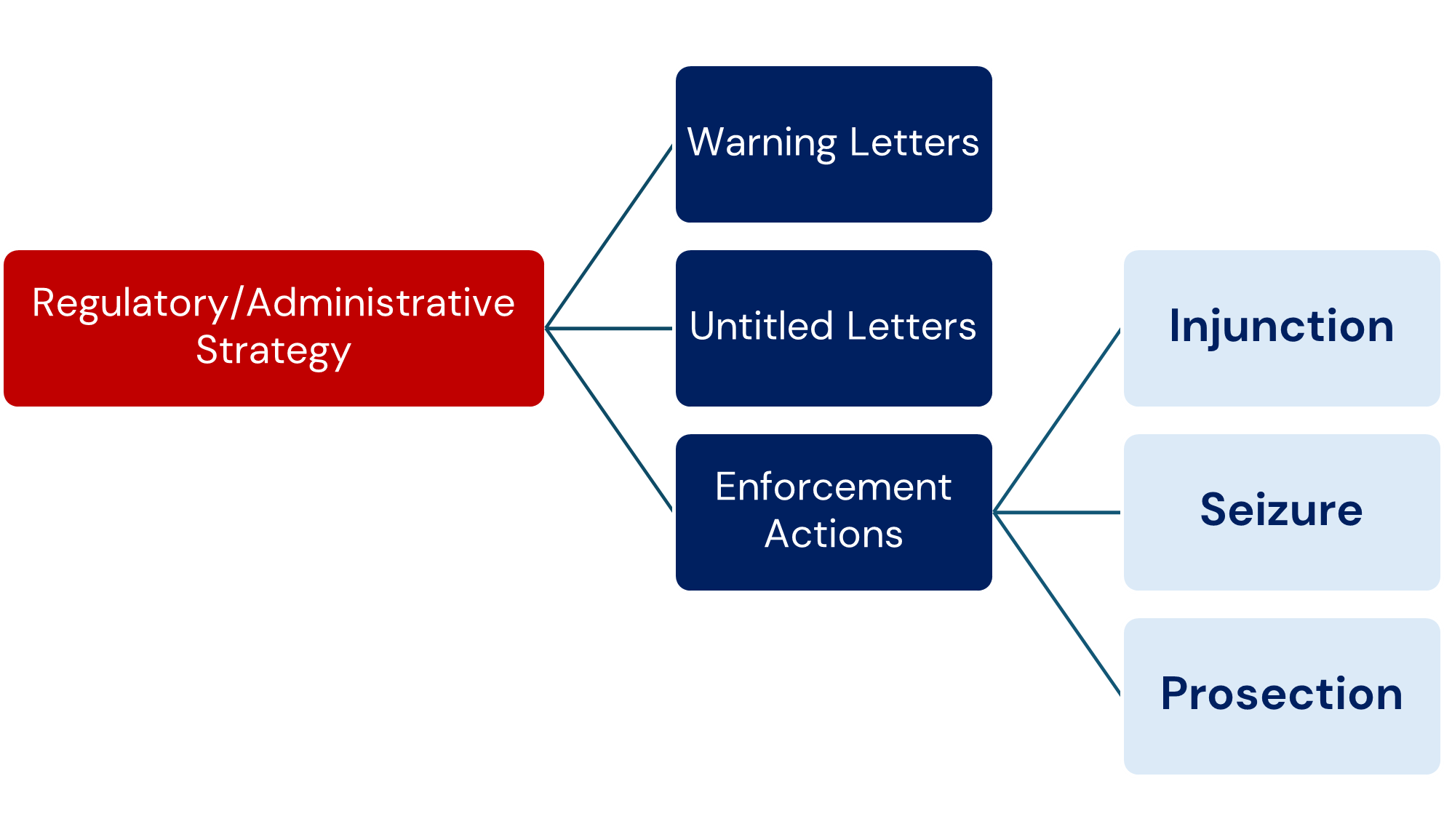
a) Warning Letters
The issuance of a Warning Letter (WL) may be warranted when the inspection uncovers significant objectionable conditions related to noncompliance with PADE requirements. The CDER PVC Team and OSI management will evaluate all inspections classified as OAI by OBIMO on a case-by-case basis.
b) Untitled Letters
An Untitled Letter (UL) may be warranted when the deficiencies found at the firm are severe enough to justify a formal letter to the firm, but do not meet the threshold of regulatory significance for a WL.
Factors that influence the issuance of a WL or UL include the nature and extent of the violations (for example, if they are repeated or deliberate), the compliance history of the inspected firm, and the corrective actions implemented by the firm.
c) Enforcement Actions
- Injunction: Injunction should be considered when follow-up inspection(s) show that the firm has a continuing pattern of significant and substantial deviations, despite previous attempts by FDA to obtain compliance. 1.
- Seizure: Seizure for failure to comply with post marketing adverse drug experience reporting regulations would be possible only if the approval of the application for the product has first been withdrawn (FD&C Act, section 304(a)(1)). Seizure would then be based on distribution of an unapproved drug product. 2.
- Prosecution: Evidence that a firm is submitting false information, not submitting required reports for serious post marketing adverse events, or withholding important information, the submission of which may have resulted in the Agency requiring labelling changes or withdrawing an application, should be referred to the Office of Criminal Investigations (OCI) for consideration of prosecution.
References
- https://www.fda.gov/
- Number of 483 issued from the System* Inspections ending between 10/1/2022 and 9/30/2023 https://www.fda.gov/inspections-compliance-enforcement-andcriminal- investigations/inspection-references/inspection-observations
- Post marketing Drug Safety Compliance: 2019 Inspection Findings April 29, 2020 (Live Webinar) Centre for Drug Evaluation and Research – Small Business and Industry Assistance, Centre for Drug Evaluation and Research, US Food and Drug Administration
- Post marketing Drug Safety and Inspection Readiness June 19, 2018 Centre for Drug Evaluation and Research (CDER) Small Business and Industry Assistance (SBIA) Webinar
- Post marketing Drug Safety and Inspection Readiness June 19, 2018 Centre for Drug Evaluation and Research (CDER) Small Business and Industry Assistance (SBIA) Webinar
- CHAPTER 53 – Post marketing Surveillance and Epidemiology: Human Drug and Therapeutic Biological Products, fda.gov
About Soterius
Soterius is a strong team of pharma professionals who design customized, innovative, and cost-efficient processes for clinical safety, pharmacovigilance, and medical affairs. Our deep industry knowledge and up to date insights let us combine agile, people powered intelligence in pioneering customer centric solutions. Our innovative technology solutions include engagement tools and communications platforms to create a unified and compliant medical access facility. With a strong global presence, we provide comprehensive clinical and post marketed safety services, that include aggregate report writing, signal detection and management, global literature surveillance, risk management, case processing and regulatory reporting. We use state-of-the-art technologies to solve complex safety operations problems, be it case processing, intake, site reporting for clinical trials, or literature search and management. We have one of the most accurate solutions for case intake and case processing using AI.
We support companies from the initial development stage of a drug/vaccine to the approval and ultimate marketing of the therapy, supporting ongoing operations and regulatory commitments globally.
Authors

Dr. Sumit Verma MD, DNB
President, Clinical Safety and PV
Dr. Sumit Verma is a medical graduate with specialization in anesthesiology and has more than 15 years of experience in the pharmaceutical industry, clinical medicine, clinical research, and pharmacovigilance. He has built teams that have consistently delivered and exceeded customer expectations across pharmacovigilance domains such as case processing, signal management, risk management, aggregate reports, and clinical safety. He has co-authored two books – one on pharmacovigilance and another on pharmacology.
Disclaimer
Copyright 2025 by Soterius, Inc. All rights reserved. Soterius logo are trademarks or registered trademarks of Soterius in all jurisdictions. Other marks may be trademarks or registered trademarks of their respective owners. The information you see, hear or read on the pages within this presentation, as well as the presentation’s form and substance, are subject to copyright protection. In no event, may you use, distribute, copy, reproduce, modify, distort, or transmit the information or any of its elements, such as text, images or concepts, without the prior written permission of Soterius. No license or right pertaining to any of these trademarks shall be granted without the written permission of Soterius (and any of its global offices and/or affiliates). Soterius reserves the right to legally enforce any infringement of its intellectual property, copyright and trademark rights. Any content presented herewith should only be considered for general informational purposes and should not be considered as specific to the requirements of any particular organisation or for any specific purpose. Soterius does not make any representations or warranties about the completeness, reliability, appropriateness, relevance, or accuracy of the content presented here.
Confidential Information
Copyright@2025
Soterius, Inc
Discover more
 abinash kumar •
10 Min Read
abinash kumar •
10 Min Read
Governance and Accountability in AI-Enabled Pharmacovigilance
Governance and accountability are foundational to the use of artificial intelligence in pharmacovigilance. AI systems do not exist outside the pharmacovigilance system in which they are deployed. Decisions about their…
 abinash kumar •
10 Min Read
abinash kumar •
10 Min Read
Human Oversight in AI-Enabled Pharmacovigilance: What ‘HITL’ Actually Has to Mean
1. Why Human Oversight Is a Control, Not a Reassurance As artificial intelligence becomes embedded in pharmacovigilance workflows, the presence of a human reviewer is often assumed to be a…
 admin •
10 Min Read
admin •
10 Min Read
Deploying an Inspection Ready AI System in PV
Artificial intelligence in pharmacovigilance should not be assessed by how advanced the technology is. It should be assessed by what happens when the technology is wrong. Executive Summary Artificial intelligence…