Introduction
EudraVigilance is a centralised European database of suspected adverse reactions to medicines that are authorised or being studied in clinical trials in the European Economic Area (EEA).
EudraVigilance is the system for managing and analysing information on suspected adverse reactions to medicines which have been authorised or being studied in clinical trials in the European Economic Area (EEA). The European Medicines Agency (EMA) operates the system on behalf of the European Union (EU) medicines regulatory network.
EudraVigilance supports safe and effective use of medicines by facilitating::
- Electronic exchange of ICSRs (Individual Case Safety Reports) between EMA, NCAs, MAHs and CT Sponsors in the EEA
- Early detection and evaluation of possible safety signals
- Better product information for medicines authorised in the EEA.
This electronic reporting is mandatory for marketing authorisation holders and sponsors of clinical trials. Marketing authorisation holders and sponsors of clinical trials must report and evaluate suspected adverse drug reactions during the development and following the marketing authorisation of medicinal products in the European Economic Area (EEA). Marketing authorisation holders must also electronically submit information on medicinal products authorised in the European Union (EU).
January 1, 2014
My Heading 1
January 1, 2015
My Heading 2
January 1, 2015
My Heading 2
January 1, 2015
My Heading 2
| Heading 2 | Heading 2 | Heading 2 |
|---|---|---|
| Sample #1 | Row 1, Content 1 | Row 1, Content 2 |
| Sample #2 | Row 2, Content 1 | Row 2, Content 2 |
| Sample #3 | Row 3, Content 1 | Row 3, Content 2 |
Why do we need a responsible Person for EudraVigilance (RPEV)
The Qualified Person for Pharmacovigilance (QPPV, for MAHs), or the Responsible Person (RP, for Sponsors/Non-Commercial Sponsors) is responsible for managing an organization and its users in the EudraVigilance Production system.

For an individual to provide RP services to a sponsor/non-commercial sponsor, any one user in the individual’s organization should successfully complete the EudraVigilance ICSR knowledge evaluation and XEVMPD knowledge evaluation. To ensure the quality of data submitted to the EMA, the EMA offers training courses on ICSR and Product submissions using EVWeb, after which users may undergo knowledge evaluations.
Registration of Responsible Person in EudraVigilance
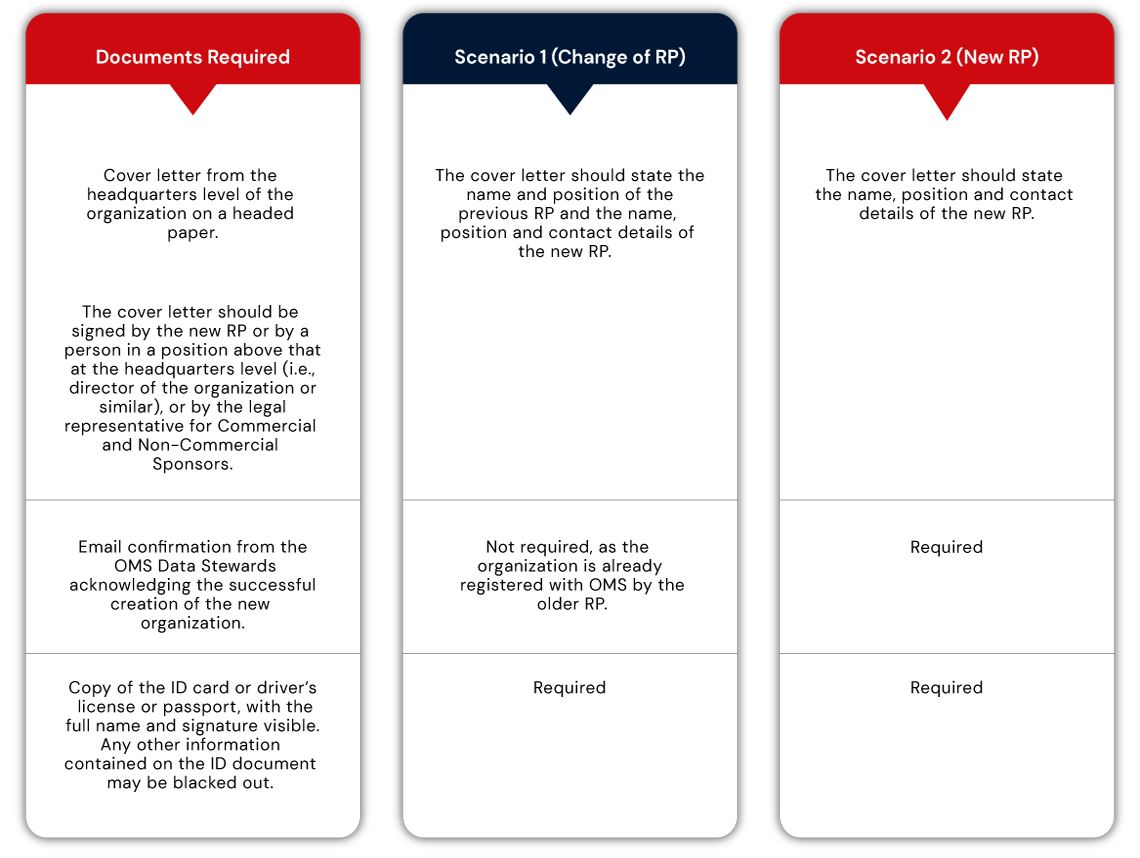
Registration of a Responsible Person could entail one of the following two scenarios:
Scenario 1: Change of RP (when there is still an RP in the organization), or
Scenario 2: New RP registration (when the first user of a new organization is registering as RP)
A set of documents must be submitted to the EMA to register the Responsible Person in either of the above scenarios.

Conclusion
We hope that this blog was helpful in understanding the role of a Responsible Person and the process of registration of a RP with the EMA.
Please reach out to us if you require RP services in the EEA, or wish to understand more about the registration process for the RP.
| References: 1. EMA EudraVigilance Registration Manual. 17 March 2022. EMA/13454/2020, Rev. 11. European Medicines Agency. 2. EudraVigilance user declaration for qualified person for pharmacovigilance/responsible person for EudraVigilance. 05 October 2020.EMA/204890/2017. European Medicines Agency. 3. New Organization First User QPPV/RP or Change of EU QPPV/RP. 29 April 2021. EMA/503895/2018. European Medicines Agency. 4. https://www.ema.europa.eu/en 5. EudraVigilance | European Medicines Agency (europa.eu) 6. Some images from Pixabay (www.pixabay.com) |
Authors

Dr. Sumit Verma – Operations, Co-Founder
Dr. Sumit Verma is a medical graduate with specialization in anesthesiology and has more than 15 years of experience in the pharmaceutical industry, clinical medicine, clinical research, and pharmacovigilance. He has built teams that have consistently delivered and exceeded customer expectations across pharmacovigilance domains such as case processing, signal management, risk management, aggregate reports, and clinical safety. He has co-authored two books – one on pharmacovigilance and another on pharmacology.
Tanvi Chaturvedi– Senior Manager, Clinical Safety and Pharmacovigilance
Tanvi is a pharma professional with more than 10 years of experience in pharmacovigilance. She has expertise in pharmacovigilance and clinical safety processes, including pre-approval, postmarketing and late phase safety requirements and has managed end-to-end project setup for EU and US based projects that include process development, safety database and medical information database setup with data migration and validation, transition of overall PV services, team selection and mentoring, and troubleshooting across PV verticals. Has experience across various domains like ICSR processing in safety databases, literature management, aggregate reporting, signal management, regulatory intelligence, EudraVigilance systems, system automation, process development and transition, and pharmacovigilance quality assurance. She is eligible to provide Responsible Person for EudraVigilance (RPEV) services to Sponsors of Clinical Trials in the EU.

Disclaimer:
Any content presented in the document should be considered for general informational purposes and should not be considered as specific to the requirements of any particular organisation or for any specific purpose. Soterius does not make any representations or warranties about the completeness, reliability, appropriateness, relevance or accuracy of the content in the document. Please refer to the relevant regulations/guidance for information.