Medication errors are an important concern in healthcare systems around the world. They can occur at any stage of the medication process, from prescribing to administration, and can have serious consequences in patients. Some examples of medication errors could include giving a medication to the wrong patient, giving the wrong dose of a medication, not prescribing a medication that was indicated, entering an order for the wrong patient, or forgetting to give a medication that was due. In this article, we will explore the definition of medication errors, the various stages where they can occur, the role of healthcare professionals in reporting and preventing these errors, and strategies for reducing their occurrence.
Defining Medication Errors
A medication error is a commonly used term and is defined as an unintended failure in the treatment process that leads to, or has the potential to lead to, harm to the patient (Ferner & Aronson, 2006; EMA Guidance 2015). These errors can take place at multiple stages of the medication-use system, such as during prescribing, entering data into computer system, preparing or dispensing the drug, or administering the drug to the patient.
Why are Medication Errors Important – Prevalence and Impact of Medication Errors
The FDA receives more than 100,000 US reports each year associated with a suspected medication error. Individual studies have reported hospital inpatient medication error rates of 4.8% to 5.3% and in another study, prescribing errors for inpatients occurred 12.3 times per 1,000 patient admissions. One study on the frequency of medication errors revealed that fewer than 1% of medication errors resulted in an adverse event.
These medication error reports come from various sources, including drug manufacturers, healthcare professionals, and consumers. These reports are reviewed by the FDA and are classified to determine the cause and type of the error. Medication errors can also result in serious consequences. In addition to the harmful impact on patients, medication errors can also impose significant costs on healthcare systems. Estimates suggest that these errors, in addition to decreasing the patients’ confidence in medical services, can cost between $6 billion and $29 billion per year in the United States alone.
Relationship between Medication Errors and Adverse Effects
The Figure below reflects the interplay between medication errors and harm (i.e. associated with adverse reaction and preventability).
Adverse reactions that result from medication errors are considered preventable, in contrast to generally non-preventable adverse reactions which are mentioned in the Product Label (e.g. in Prescribing Information, SmPC etc.) for which the chances of adverse event occurrence is usually known and accepted and will likely occur depending on the frequency of the adverse reaction and on other circumstances such as concomitant medication use, underlying disease condition etc.
There are medication errors which do not essentially result in adverse reaction, but which are important from their unwanted effects e.g. from an economic or environmental standpoint.
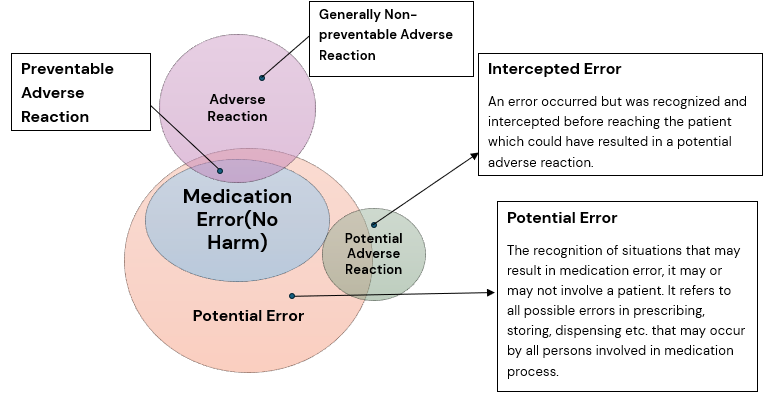
Figure 1: Figure: Correlation between medication errors, preventable and generally non-preventable adverse reactions and intercepted errors (modified according to Morimoto et al., Qual Saf Health Care 2004; 13:306-314). Ref: EMA Good Practice Guide on recording, reporting & assessment of medication errors; 23 Oct 2015.
Stages of Medication Errors
Medication errors can occur at various stages of the drug delivery process. These stages include:
-
Prescribing
Errors during this phase can occur due to incorrect drug selection, dosage, or frequency, or by failing to consider the patient’s medical history or potential drug interaction.
-
Transcribing
Errors can occur when entering prescription information into a computer system, such as incorrect drug selection, dosage, or frequency.
-
Dispensing
Errors can happen during the preparation and distribution of the drug, such as dispensing the wrong medication, incorrect dosage, or improper labelling.
-
Administration
Errors can occur when administering the drug to the patient, such as administering the wrong drug, dose, or route, or at the wrong time
-
Monitoring
Errors can happen when healthcare professionals fail to monitor the patient’s response to the medication, leading to potential adverse reactions or failure to achieve therapeutic goals.
Recording and Reporting Medication Errors
Medication errors associated with drug use may be spontaneously reported as unsolicited reports by a consumer, healthcare professional or marketing authorization holder, or may be reported as solicited reports of suspected adverse reactions from data collection systems e.g. non-interventional studies or registries.
Suspected (serious and non-serious) adverse reactions associated with medication errors should be recorded, reported and assessed. The reports of medication errors not associated with adverse reaction should also be recorded; however, these cases are not reportable as valid ICSR.
The marketing authorization holders should summarize relevant information on medication errors, even when not associated with adverse outcomes, in the periodic safety update reports and the risk management plans. The figure below provides a summary of recording of medication errors from the perspective of pharmacovigilance and patient safety.
| Category |
Medication Error with AR |
Medication Error without AR |
Intercepted Error |
Situations Capable of Causing Medication Error |
| Medication Error |
Yes |
Yes |
Yes |
No |
| Adverse Reaction (AR) |
Yes |
No |
Not Applicable |
Not Applicable |
| Recording of Medication Error |
Medication Error with AR |
Medication Error without AR |
Intercepted Medication Error |
Situations capable of causing Medication Error |
| Report Type / Relevance |
Record ICSR Report, if applicable PSUR, RMP |
PSUR, RMP |
PSUR, RMP |
PSUR, RMP |
Strategies for Reducing Medication Errors
Various mechanisms can be employed to reduce the occurrence of medication errors.
The FDA is actively involved in efforts to prevent medication errors. Before approving drugs for marketing, FDA reviews the drug name, labelling, packaging, and product design to identify and revise information that may contribute to errors. FDA reviews the following to reduce the incidence of medication errors:
- Proposed brand names to minimize confusion among drug names. Look-alike and sound-alike names are to be avoided.
- Container labels to ensure healthcare providers and patients can select the correct drug and easily differentiate between different strengths of the same drug.
- Prescribing and patient information to ensure clear and easy-to-read instructions for use.
After the drugs are approved for marketing, FDA continues to monitor and evaluate medication error reports.
- FDA may require manufacturers to revise labels, packaging, product design, or proprietary names to prevent errors.
- FDA may also issue communications to alert the public about medication error safety issues.
- FDA collaborates with regulators, external stakeholders, standard-setting organizations, patient safety organizations, and researchers to understand the causes of medication errors and develop interventions to prevent them.
Implementing Barcodes on Drug Labels
The FDA has introduced rules requiring barcodes on certain drug and biological product labels. These barcodes allow healthcare professionals to use barcode scanning equipment to verify that the correct drug, dose, and route of administration are being given to the right patient at the right time, to reduce medication errors in hospitals and other healthcare settings.
Providing Guidance to Manufacturers
The FDA has published guidance documents to help manufacturers design drug labels, packaging, and select drug names in a way that reduces or eliminates hazards contributing to medication errors. These guidance documents offer recommendations on various aspects of drug design, such as imprint codes on tablets, appropriate dosing devices, and package design to protect consumers against incorrect use.
Importance of Consumers in Reducing Medication Errors
Consumers play a crucial role in reducing medication errors. Some tips for the consumers to minimize the risk of medication errors include the following:
- Be aware of the various causes and risks of medication errors.
- Know the name and purpose of the medications that is prescribed.
- Know how to take the medication and follow the directions properly including directions for storage.
- Check the container’s label every time the medication is taken.
- Keep medications stored in their original containers.
- Maintain an updated list of all medications, including over-the-counter drugs, supplements, and other substances and share this list with your healthcare provider.
- Be aware of the risk of drug-drug and drug-food interactions.
- Ask the pharmacist or healthcare provider in case of any questions about the medication.
- Report suspected medication errors.
Specific Consideration of Medication Errors in High-Risk Groups
Consumers play a crucial role in reducing medication errors. Some tips for the consumers to minimize the risk of medication errors include the following:
-
Paediatric patients
Paediatric patients may be at especially high risk of medication errors, with dosing errors being the most common type of error. This may be due to the variation in age, size and weight, and body surface area in children. Overdose was the most commonly reported medication error (accounting for 21% of all reports) in a study of paediatric patients while underdosing was the most commonly reported medication error in certain paediatric specialties in another study. These conflicting findings suggests a more general risk of dosing errors (leading to either over- or underdosing) in paediatric patients.
Paediatric prescribing is often decided by the patient’s weight, yet weight is not measured before each prescription and can change over time. Further, mathematical miscalculations may also occur. Accidental ingestion or other unintended use of medicinal products by children should be prevented. A standard statement that drugs should be kept out of the sight and reach of all children is included on the labelling for all products and use of child-resistant packaging may also be considered.
-
Elderly patients
Elderly patients are at a high risk of medication errors. These patients may have physical and cognitive impairment and therefore may have difficulties in taking medicines, e.g. swallowing tablets, opening packaging or reading user instruction. Moreover, elderly patients frequently use multiple drugs (polypharmacy) which in itself may cause compliance problems which may be partly overcome by the design of the drugs (e.g. a wider range of colours, sizes and tablet shapes is known to assist the recognition of medicines).
Elderly patients are more likely to experience impaired swallowing. This may result in accidental underdosing, which can be appropriately managed by the use of formulations which are easier to swallow. It is important that appropriate materials for elderly patients are developed, including use of large print text and Braille for patients with impaired eyesight. Older people may also more frequently require the assistance of caregivers. The caregiver, nurse and family can play an important role for the correct use of the medicines and should be proactively involved by the doctor or pharmacist.
Conclusion
Medication errors are a significant concern in healthcare systems worldwide and is an important cause of patient morbidity and mortality. Medication errors can occur due to varied factors such as medication factors (e.g. similar sounding names, low therapeutic index), patient factors (e.g. poor renal or hepatic function, impaired cognition, polypharmacy), and health care professional factors (e.g. use of abbreviations, cognitive biases) which can precipitate medication errors. With the combined efforts of the FDA, healthcare professionals, manufacturers, and consumers, it can be ensured that the medication-use process is safer and more effective for all involved.