What is a Safety Signal in Pharmacovigilance?
Information arising from one or multiple sources, including observations and experiments, which suggests a new potentially causal association, or a new aspect of a known association between an intervention and an event or set of related events, either adverse or beneficial, that is judged to be of sufficient likelihood to justify verificatory action. New aspects of a known association may include changes in the frequency, distribution (e.g. gender, age and country), duration, severity or outcome of the adverse reaction. A signal often relates to all medicinal products containing the same active substance, including combination products. Certain signals may only be relevant for a particular medicinal product or in a specific indication, strength, pharmaceutical form or route of administration whereas some signals may apply to a whole class of medicinal products.
Signal Management – What it is:
A set of activities performed to determine whether, based on an examination of individual case safety reports (ICSRs), aggregated data from active surveillance systems or studies, scientific literature information or other data sources, there are new risks associated with an active substance or a medicinal product or whether known risks have changed, as well as any related recommendations, decisions, communications and tracking.
What do the regulators say:
- 21CFR312.32
Review of safety information:
The sponsor must promptly review all information relevant to the safety of the drug obtained or otherwise received by the sponsor from foreign or domestic sources, including
information derived from any clinical or epidemiological investigations, animal or in vitro studies, reports in the scientific literature, and unpublished scientific papers, as well as reports from foreign regulatory authorities and reports of foreign commercial marketing experience for drugs that are not marketed in the United States.
- USFDA draft guidance on sponsor responsibilities – safety reporting requirements and safety assessment for IND and Biovailability / Bioequivalence studies
Systematic Approach for Review of Safety Information (§ 312.32(b)):
Sponsors should have a systematic approach to safety surveillance to comply with the IND safety reporting requirements and to improve the overall quality of safety reporting. Such an approach should include a process for promptly reviewing, evaluating, and managing accumulating data on SAEs from the entire drug development program that are sent from domestic or foreign sources. During the course of drug development, investigators who conduct clinical trials generally report to the sponsor adverse event information; however, a sponsor may become aware of new safety information from a variety of sources, both domestic and foreign. Compliance with post marketing safety laws and regulations for human drugs and therapeutic biologics.
The sponsor must review and evaluate safety information from any source regardless of whether the data came from studies conducted under the IND (§ 312.32(c)(1)(ii) and (iii)) to determine if there is a newly identified significant risk to trial participants. Sources include but are not limited to:
- Animal or in Vitro Studies
- Clinical or epidemiological investigations
- Reports in the scientific literature, including unpublished reports of which the sponsor becomes aware
- Information presented at professional or scientific meetings (e.g. abstracts)
- Reports from foreign regulatory authorities
- Reports from commercial marketing experience, including outside the United States
- European Medicine Agency (EMA)
Safety surveillance: The sponsor along with the investigators is responsible for the ongoing safety
surveillance and risk minimization of the study subjects during the clinical study duration, taking, if required, appropriate urgent safety measures for protection of study subjects. The Member State Concerned will monitor the risks of investigational medicinal products considering the available information according to the procedures laid down in the implementing Regulation on rules and procedures for the cooperation of the Member States in safety assessment of clinical trials. Safety reporting with regard to authorised Auxiliary Medicinal Products shall be made in accordance with the normal safety reporting requirements
for authorised medicinal products.
The notifications required from sponsor or investigator in relation to safety of the clinical study subjects include:
- Reporting of adverse events and serious adverse events
- Reporting of suspected unexpected serious adverse reactions (SUSARs)
- Other unexpected events relevant for the subject’s safety
- Annual safety report
- Reporting of serious breaches
- Urgent safety measures
- Temporary halt of the study due to safety reasons
- CIOMS Working Group VI
Identification and Evaluation of Risk from Clinical Trial Data – Ongoing Safety Evaluation: The purpose of ongoing safety evaluation during drug development is to ensure that important safety signals are detected early and to gain a better understanding of the benefit-risk profile of the study drug.
- Clinical trial sponsors should develop a system to analyse, evaluate and take actions on the safety information received during drug development on a continuous basis. This is to ensure the earliest possible detection of safety concerns and allow suitable risk minimization, such as of ongoing study protocol revisions, to ensure that clinical trial participants are not exposed to undue risk.
- Safety monitoring, evaluation and analysis should be performed in such a way so that the integrity of the individual studies or the overall development program is not compromised. Study sponsor should be totally aware of the potential risks of the investigational product and the morbidities characteristic of the study population at every stage of drug development.
How Soterius can help you in Setting Up Signal Detection Process:
-
Team of Experienced Physicians and Product Specific Trained Resources
– The signal management team adheres to a process involving a thorough review and medical evaluation of safety data.
-
Safety Surveillance Plan
– The components of the safety surveillance plan include the following:
| Data Collection from
Various Sources |
Preparation of Detailed
Signal Evaluation Reports |
Signal Report Discussion
in Safety Review Meeting |
| · Safety database line listings, Clinical database line listings including laboratory investigations
· Published literature
· Aggregate reports/Risk management plans
· Regulatory Websites depending on authorization of client’s products in various countries/regions e.g.
· FDA website*
*(Potential Signals of Serious Risks/New Safety Information Identified from FAERS) |
· Sources of signal detection Identified drug-event pairs
· Signal validation Other relevant safety issues |
· Discussion with key stakeholders prior to finalization on periodic basis
· Further actions (Routine monitoring, Potential Signals Y/N, Additional data search) |
Signal Management Process:
Signal detection, Signal validation, Signal analysis and prioritization, Signal assessment, and Recommendations for action.
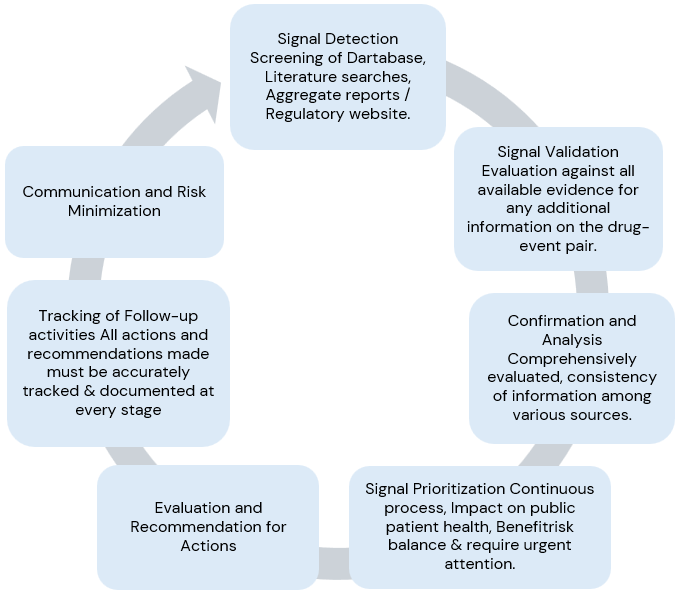
In conclusion, signal management is a process that requires a high-level standard operating procedure (SOP) describing:
- How signal prioritization and evaluation are approached (e.g. what does the signal prioritization imply?
- How are the sources of safety data queried? Who performs the signal evaluation?).
- How is the risk determination performed (e.g. what criteria have been considered and what data are available to qualify the risks?).
- How is the best course of action determined?
- How, when and to whom the potential or identified risks are communicated.
Once a signal is identified, the following steps are necessary:
- Assess public health impact.
- Validate and assess the strength of the signal.
- Define data sources and limitations.
- Compile safety data.
- Compare data from different sources.
- Evaluate characteristics and likelihood of the risk associated with the signal.
- Determine appropriate actions for evaluation, communication, and risk reduction.
Remember…. if the information available suggests that there could be a risk that requires prevention or minimization in a timely manner
- Always use clinical judgement and flexibility throughout the process.
- Be prepared to take action at any stage before the formal signal assessment is completed.
- Points for consideration: severity, seriousness, outcome, reversibility, exposure in vulnerable populations, expected extent of regulatory intervention, consequences of treatment discontinuation & availability of other therapeutic options etc.
This was a small start with a quick reference to importance of Signal detection in Early Phase Clinical Development Programs. I would be discussing various aspects of signal management process in the upcoming articles.