
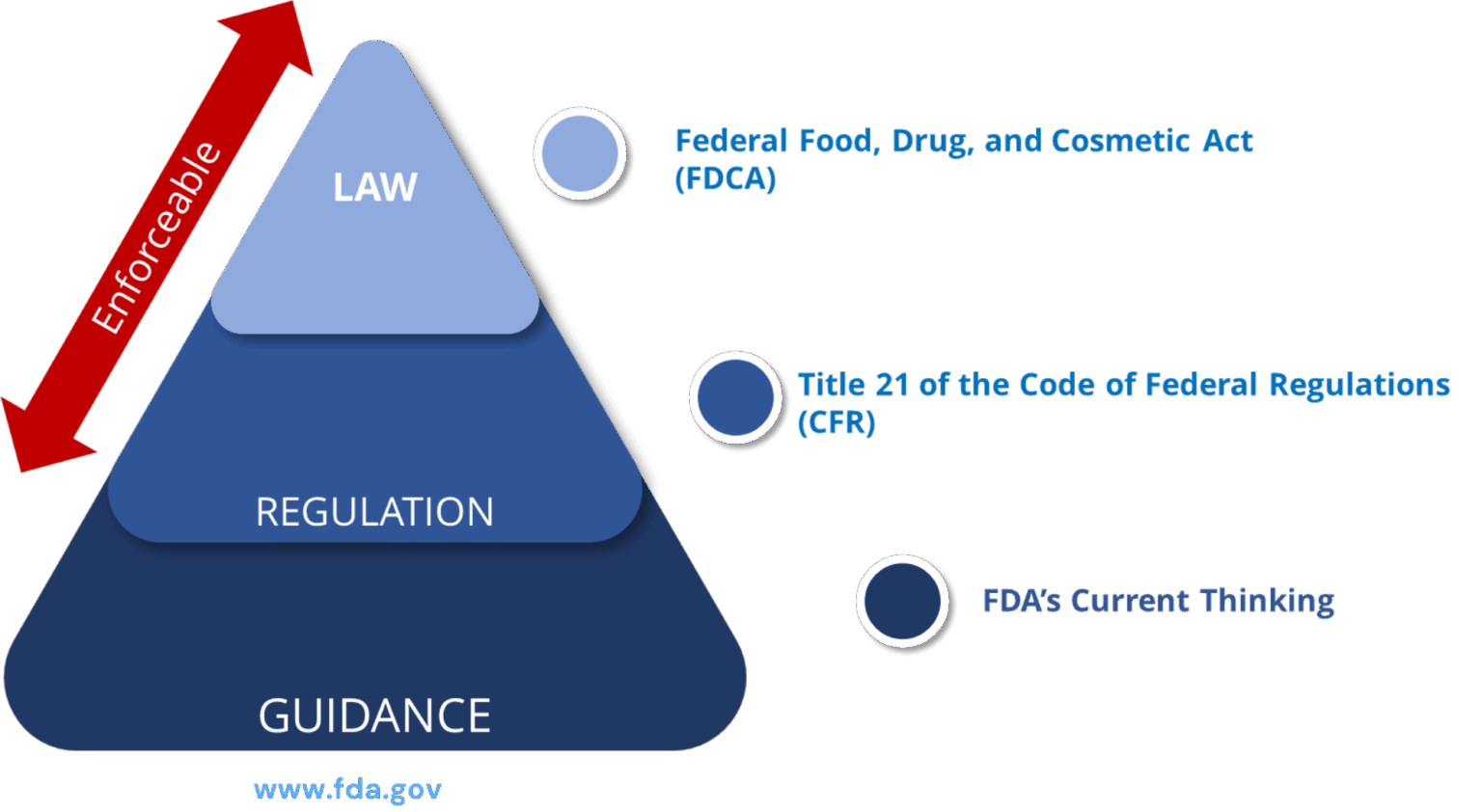
Legal Framework of PADE Inspections
Good Corrective Action Plan – Four Reasons to Submit a Complete and Timely
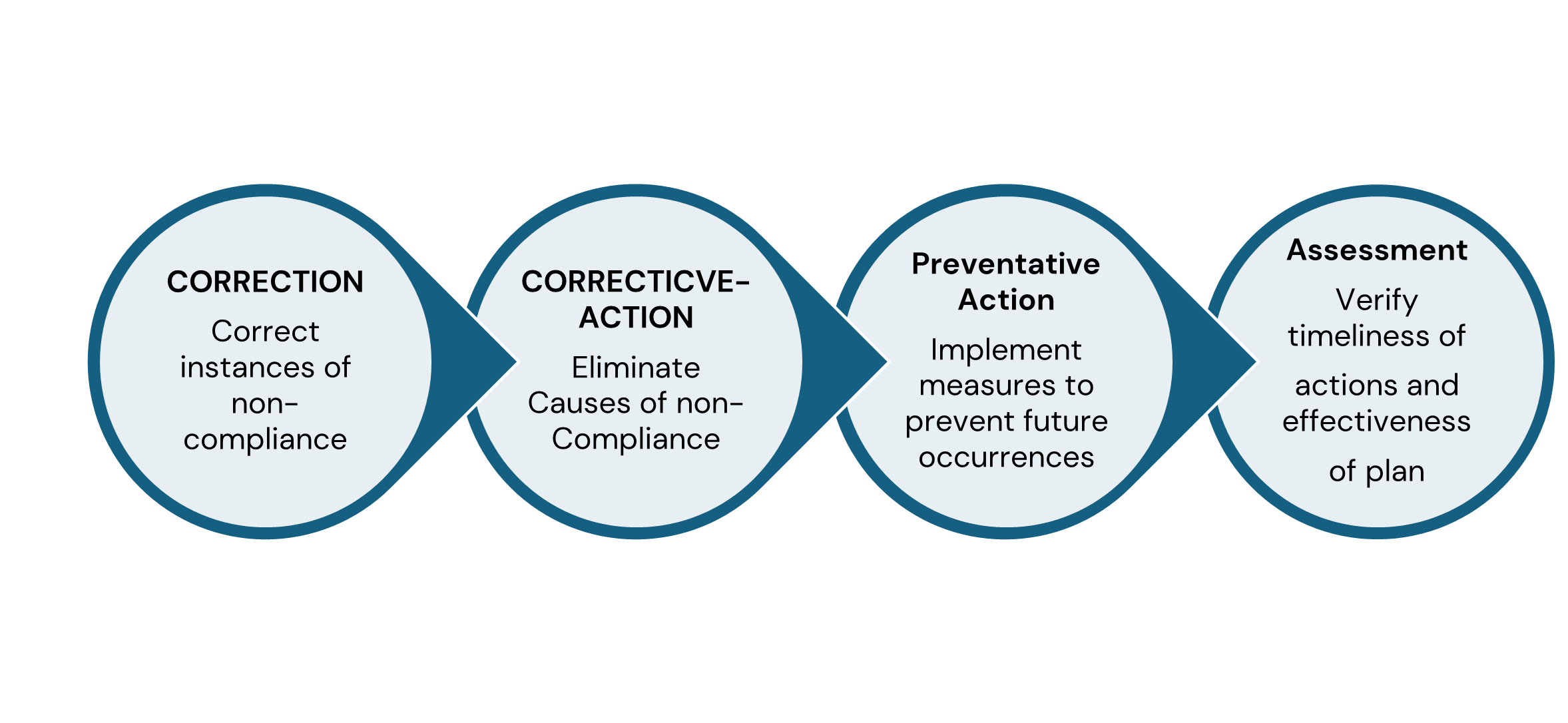
Written Response
- May be considered in an FDA compliance decision.
- Demonstrates your acknowledgment and understanding of the observations to the FDA
- Demonstrates your commitment to correct the observations to the FDA
- Establishes credibility with the FDA
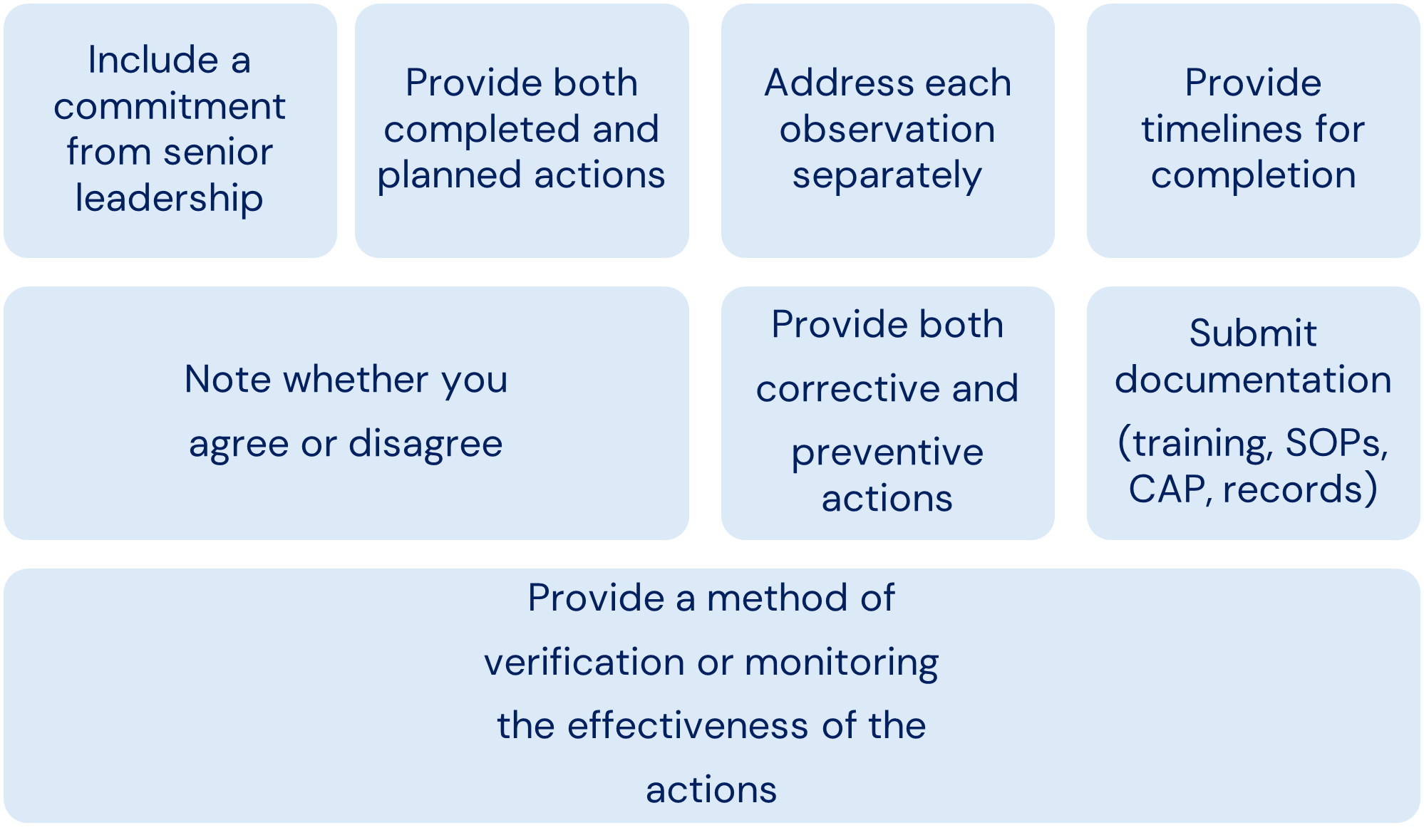
Points to Consider for Written Responses to the FDA
(SUBMIT THE REPORT WITHIN 15 WORKING DAYS)
Inspection Reporting: FORM FDA 483, Inspectional Observations

Inspection Classifications
| No Actions Indicated
(NAI) |
Voluntary Action Indicated (VAI) | Official Action Indicated (OAI) |
| No Objectionable conditions
or practices were found during an inspection (or the objectionable conditions found do not justify further regulatory action).
|
Objectionable conditions or practices were found, but do not rise to the level warranting OAI classification. | Objectionable conditions or practices were found, whose scope, severity, or pattern warrants the recommendation for a regulatory action. |
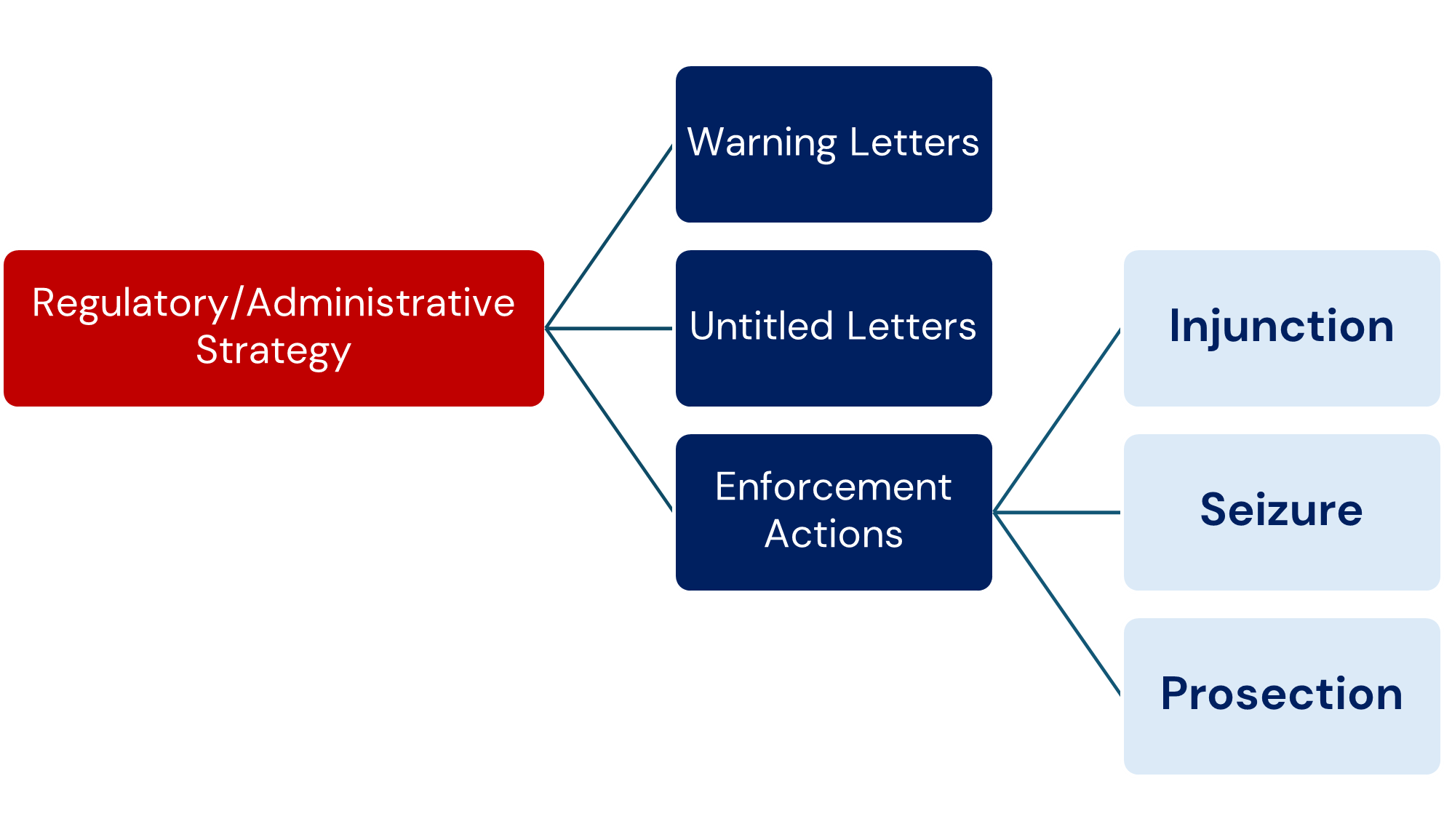
a) Warning Letters
The issuance of a Warning Letter (WL) may be warranted when the inspection uncovers significant objectionable conditions related to noncompliance with PADE requirements. The CDER PVC Team and OSI management will evaluate all inspections classified as OAI by OBIMO on a case-by-case basis.
b) Untitled Letters
An Untitled Letter (UL) may be warranted when the deficiencies found at the firm are severe enough to justify a formal letter to the firm, but do not meet the threshold of regulatory significance for a WL.
Factors that influence the issuance of a WL or UL include the nature and extent of the violations (for example, if they are repeated or deliberate), the compliance history of the inspected firm, and the corrective actions implemented by the firm.
c) Enforcement Actions
- Injunction: Injunction should be considered when follow-up inspection(s) show that the firm has a continuing pattern of significant and substantial deviations, despite previous attempts by FDA to obtain compliance. 1.
- Seizure: Seizure for failure to comply with post marketing adverse drug experience reporting regulations would be possible only if the approval of the application for the product has first been withdrawn (FD&C Act, section 304(a)(1)). Seizure would then be based on distribution of an unapproved drug product. 2.
- Prosecution: Evidence that a firm is submitting false information, not submitting required reports for serious post marketing adverse events, or withholding important information, the submission of which may have resulted in the Agency requiring labelling changes or withdrawing an application, should be referred to the Office of Criminal Investigations (OCI) for consideration of prosecution.

