1. Why Human Oversight Is a Control, Not a Reassurance
- As artificial intelligence becomes embedded in pharmacovigilance workflows, the presence of a human reviewer is often assumed to be a safeguard. The implicit belief is that if a person is
involved somewhere in the process, risk is automatically controlled. That assumption is flawed. - Human oversight in AI-enabled pharmacovigilance is not a reassurance mechanism. It is a control. Its purpose is to mitigate known and foreseeable risks associated with automated
outputs, including missed information, biased interpretation, and over-reliance on system recommendations. - In safety-critical activities, simply adding a human step does not guarantee meaningful oversight. If the human role is poorly defined, lacks authority, or is limited to confirming system
outputs, the risk profile remains largely unchanged. In some cases, it may worsen, as apparent human involvement can increase unwarranted trust in the system. - Effective oversight must therefore be intentional. It requires clarity on when human judgement is expected to intervene, what decisions a human can override, and how that intervention materially affects outcomes. Without this clarity, “human review” becomes a procedural label rather than a genuine risk control.
- For pharmacovigilance organisations, this distinction matters. Oversight exists to manage the consequences of error, not to create comfort that a process appears compliant. Treating human involvement as a checkbox undermines its purpose and leaves critical risks unaddressed.
2. What Human Oversight Actually Means in Practice
- Human oversight in AI-enabled pharmacovigilance is not a single concept. It is best understood through three distinct forms of human involvement, each serving a different control purpose
1. Human-in-the-Loop (HITL):
A qualified human reviews AI outputs and has the authority to accept, modify, or reject them before they influence decisions.
2. Human-on-the-Loop (HOTL):
Humans supervise system performance over time, monitor outputs and trends, and intervene when predefined thresholds or anomalies are detected.
3. Human-in-Command (HIC):
Humans retain authority to decide whether, when, and how an AI system is used, including the ability to limit, suspend, or discontinue its use.
- These are not interchangeable labels. They represent different oversight mechanisms with different implications for risk control and accountability.
- Human-in-the-loop is most appropriate where incorrect outputs could directly affect regulatory reporting, safety assessment, or patient protection. The defining feature is not the presence of a human, but the requirement that human judgement actively determines the outcome before decisions are finalized.
- Human-on-the-loop applies where AI systems operate with a lower level of immediate decision impact. In this model, humans do not intervene in every individual output but remain
responsible for supervising performance, detecting drift, and escalating issues when behaviour deviates from expectations. This form of oversight is only effective when monitoring is robust
and escalation pathways are clear. - Human-in-command underpins all other forms of oversight. It establishes that responsibility for AI use sits with the organization, not the system. This includes decisions about scope of use,
acceptable risk, and when continued operation is no longer justified. Without this authority, other oversight mechanisms lack enforceability. - Selecting the appropriate form of oversight depends on how much influence the AI system has on pharmacovigilance decisions and the consequences of error. Choosing an oversight model that is misaligned with actual system impact can create a false sense of control while leaving material risks unmanaged.
- For Heads of Safety, the key question is not whether one of these oversight models exists, but whether the chosen model is appropriate for the use case and defensible given its impact.
Black Swan” Events in AI
A “black swan” event refers to a rare and unexpected failure with disproportionately high impact.
In pharmacovigilance, even infrequent AI errors can matter if consequences affect safety or compliance, which is why average performance metrics alone are insufficient
3. Matching Oversight Models to Pharmacovigilance Use Cases
- Selecting an oversight model is not a theoretical exercise. It must be driven by how an AI system is actually used within pharmacovigilance workflows and by the consequences of error if that
system fails. In practice, many AI-enabled pharmacovigilance activities influence decisions more directly than initially assumed. - Case processing is a clear example. AI systems used to extract medical concepts, identify seriousness criteria, or prioritize cases can affect reporting timelines, downstream assessments,
and escalation decisions. Where such influence exists, human-in-the-loop oversight is typically required, as it ensures that qualified human judgement actively determines outcomes before
regulatory or safety-relevant decisions are finalized. - Other use cases may require a different oversight approach, but only where the impact of error is demonstrably low. Examples may include AI systems used for workload balancing or visualization of trends, where outputs do not directly determine regulatory actions on a case-by-case basis.In these scenarios, human-on-the-loop oversight may be appropriate only where errors do not directly influence regulatory or safety decisions. Even then, this approach is acceptable only if
system performance is continuously monitored, deviations are readily detectable, and clear escalation and intervention mechanisms are in place. The absence of human intervention at the
level of individual outputs must be explicitly justified through risk assessment and supported by strong supervisory controls. - Across all use cases, human-in-command oversight remains non-negotiable. The organisation must retain authority over whether an AI system is used at all, under what conditions, and with
what limitations. This includes decisions to restrict scope, pause operation, or discontinue use when risks outweigh benefits. Human-in-command establishes accountability at the organisational level and ensures that oversight is enforceable rather than symbolic. - A common failure is selecting an oversight model based on intended use rather than actual operational behaviour. Outputs that are reused downstream, inform prioritisation, or influence
multiple steps in the workflow can elevate risk over time. Oversight models must therefore be revisited as workflows evolve and reliance on AI increases. - For Heads of Safety, the key principle is alignment. Oversight must match real system influence, not convenience or vendor positioning. When oversight models are proportionate to impact,
they function as genuine controls. When they are not, they create a false sense of security.
4. Why Human Review Alone Is Not Sufficient
- Including a human review step in an AI-enabled workflow does not, by itself, ensure effective oversight. In practice, human review can fail to mitigate risk if it is not deliberately designed to
counter known behavioural effects associated with automated systems. - One of the most significant risks is automation bias. When AI systems perform well most of the time, reviewers may unconsciously defer to their outputs, even when information is incomplete, ambiguous, or inconsistent. Over time, this deference can reduce critical scrutiny and lead to missed errors, particularly in high-volume environments where efficiency pressures exist.
- This risk is not eliminated simply by requiring a human to “check” outputs. If the reviewer’s role is limited to confirmation, lacks clear authority to challenge results, or is constrained by time or tooling, the presence of human review provides little additional protection. In such cases, human involvement can create the appearance of control without materially changing outcomes.
Human vs Machine Roles
Machines should perform tasks they can execute reliably and at scale.
Humans should retain tasks requiring judgement, context, and accountability.
- Effective oversight therefore requires more than procedural review. It requires clarity on what the human reviewer is expected to evaluate, when escalation is required, and how
disagreement with AI outputs is resolved. Reviewers must be supported with appropriate training, decision authority, and system transparency to enable meaningful intervention. - For pharmacovigilance organisations, this has a practical implication. Oversight models must be designed with an understanding of how humans actually interact with AI systems in real
workflows. Controls that do not account for automation bias risk becoming ineffective over time, even if they appear robust on paper. - Human oversight is intended to reduce risk, not to legitimise automation. Where human review is treated as a formality rather than an active control, it fails to serve its purpose and can leave
critical risks unmanaged.
5. Leadership Accountability and Lifecycle Oversight
- Effective human oversight of AI-enabled pharmacovigilance systems cannot be delegated solely to technical teams or embedded within operational procedures. Decisions about how AI is used, controlled, and monitored are leadership responsibilities.
- Senior pharmacovigilance leadership must understand how AI systems are deployed in practice, how their outputs influence decisions, and where errors could have impact. Reliance on vendor descriptions, average performance metrics, or intended-use statements is insufficient if they do not reflect actual workflow behaviour and decision influence.
- Oversight is not a one-time design decision. AI systems evolve, workflows change, and reliance can increase gradually over time. As systems become more embedded, their influence on safety
and regulatory outcomes may expand beyond the original scope. Leadership oversight must therefore extend across the full lifecycle of AI use, from initial risk assessment and deployment
through ongoing monitoring, change management, and, where necessary, decommissioning. - This lifecycle perspective also applies to human oversight models. A model that is appropriate at introduction may become insufficient as system performance improves, volumes increase, or
outputs are reused downstream. Regular reassessment is required to ensure that oversight remains proportionate to actual risk and influence. - Accountability for AI use in pharmacovigilance ultimately rests with the organisation. Human-in-
command oversight ensures that responsibility for risk acceptance, escalation, and continued use remains clearly assigned and enforceable. Without this, other oversight mechanisms lack
authority and effectiveness. - For Heads of Safety, the implication is clear. AI does not reduce responsibility by automating tasks. As its role within pharmacovigilance systems expands, leadership accountability becomes
more concentrated, not less.





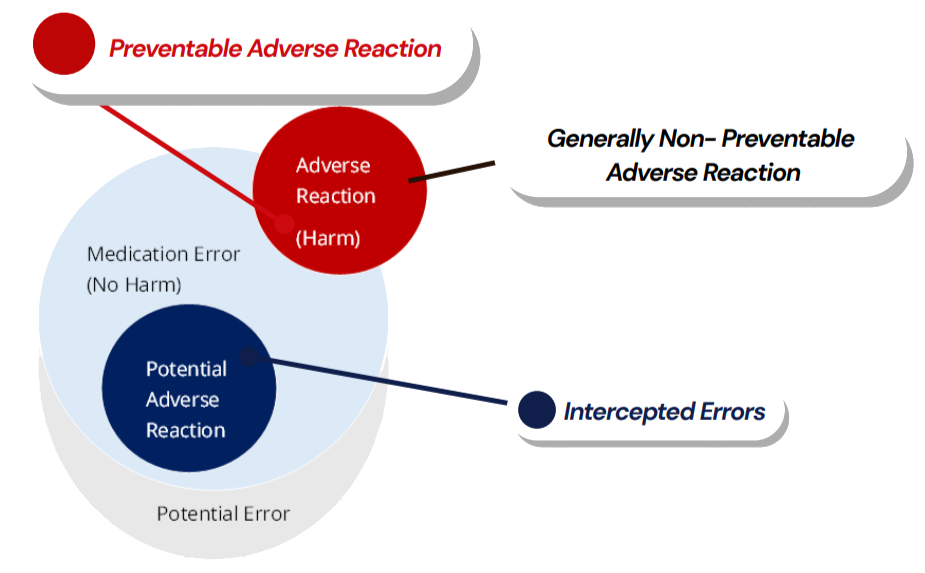

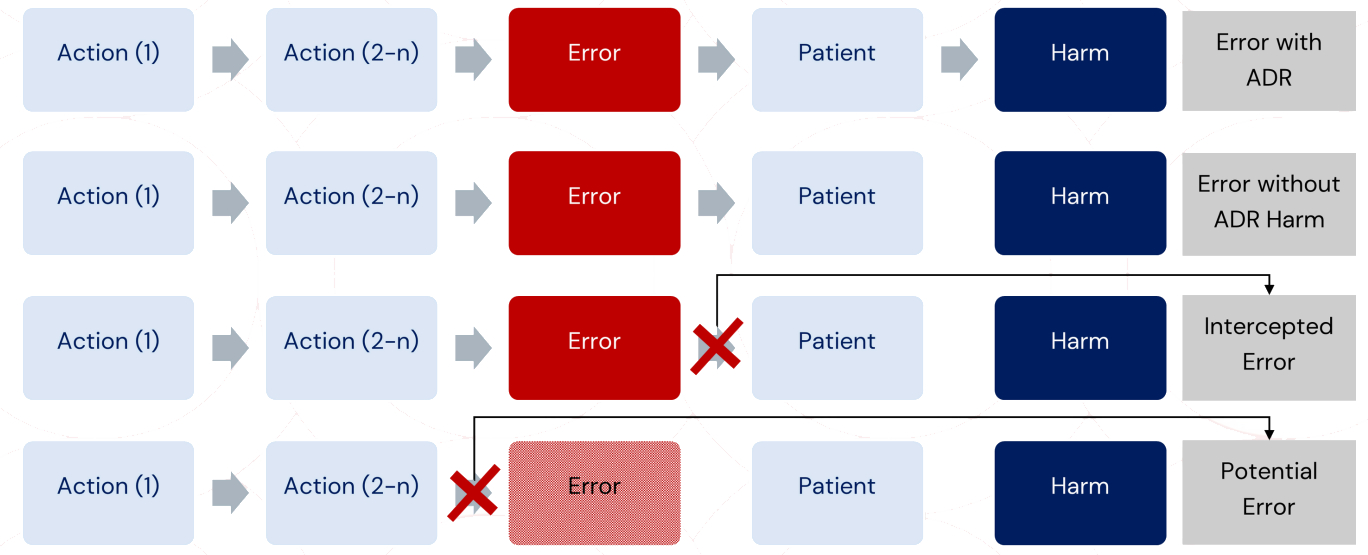 Ref: European Medicines Agency Good practice guide on recording, coding, reporting and assessment of medication errors. EMA/762563/2014
Ref: European Medicines Agency Good practice guide on recording, coding, reporting and assessment of medication errors. EMA/762563/2014